HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
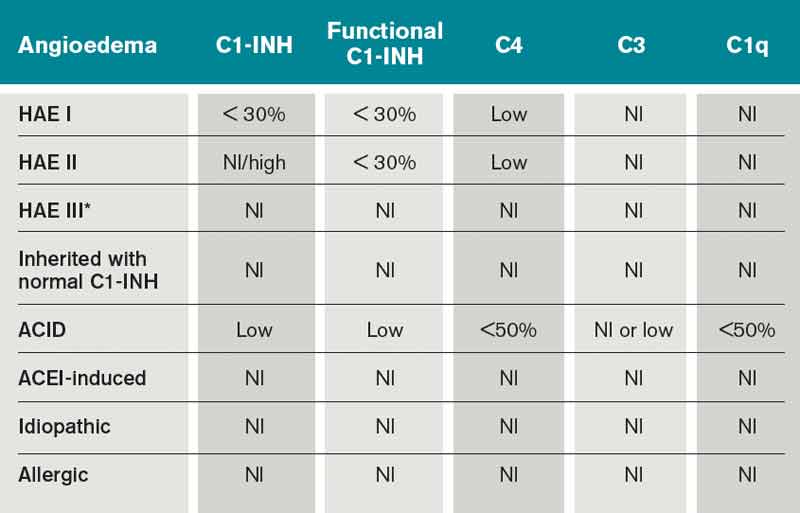
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
click for large version
Table 1. Diagnostic laboratory studies to differentiate the types of angioedema6,7
click for large version
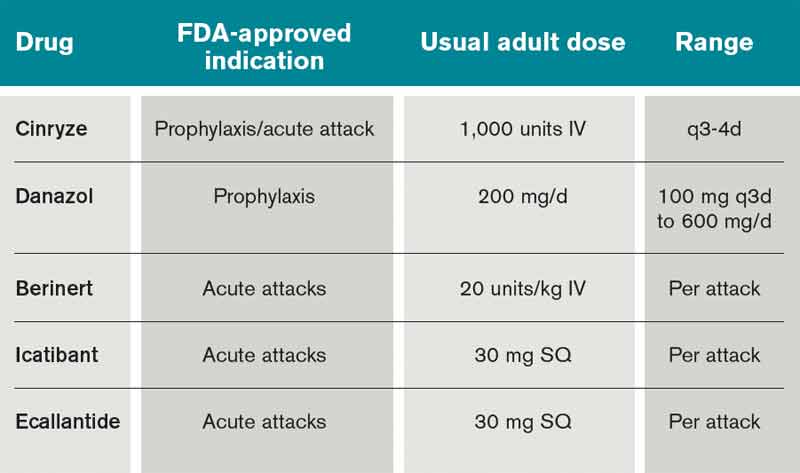
Table 2. FDA-approved available treatments for hereditary angioedema2,20
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11