click for large version
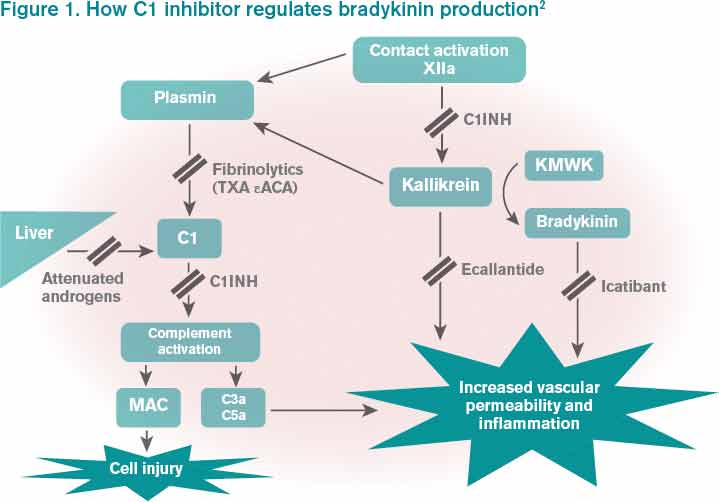
Figure 1. How C1 inhibitor regulates bradykinin production2
Case
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
click for large version
Figure 1. How C1 inhibitor regulates bradykinin production2
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3