click for large version
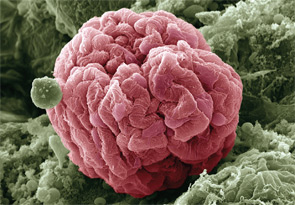
Podocyte cells, which make up the surface of the glomerulus, are pink in a 650x-magnification SEM. Glomerluar diseases can result from an acute illness, such as an upper respiratory infection, or chronic disease states, such as HIV.
The Case
A 52-year-old man presents with abdominal pain. His temperature is 100.8°F, his blood pressure is 170/90 mm/Hg, and his pulse is 110 beats per minute. On exam, he has 2+ lower extremity edema, periorbital edema, and left-sided flank tenderness. His BUN is 42 mg/dL, his creatinine is 2.5 mg/dL, and his albumin is 1.4 g/dL. Urinalysis shows 2+ protein, large blood, and red blood cells (RBCs). What are the next steps in his diagnosis?
Overview
Glomerular diseases involve a wide spectrum of disease processes. They can result from an acute illness, such as an upper respiratory infection that self-resolves, or from chronic disease states, such as HIV. In some instances, such illnesses as systemic lupus erythematosus (SLE) can cause rapidly progressive renal failure, requiring prompt intervention. While glomerular diseases can be daunting, it is essential for hospitalists to be familiar with fundamental concepts and key features unique to each syndrome.
The approach to glomerulonephritis (GN) can be simplified by summarizing various types into the two broad categories of nephrotic and nephritic syndromes, and identifying the key clinical findings (see Table 1, p. below).
The major subtypes of nephrotic syndrome are minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN), and membranoproliferative glomerulonephritis (MPGN). The clinical manifestations of nephrotic syndrome are edema, hyperlipidemia, lipiduria, and hypoalbuminemia.1 The urinalysis is significant for >3.5 g/day of proteinuria showing fatty casts or oval fat bodies.2 The loss of other proteins, such as anti-thrombin III, may put patients at higher risk for developing venous thromboses.1
The major subtypes of nephritic syndrome are post-streptococcal glomerulonephritis (PSGS), IgA nephropathy, Henoch-Schonlein Purpura (HSP), and rapidly progressive GN (RPGN types I, II, and III). The clinical manifestations of nephritic syndrome are hypertension (HTN) and hematuria.1 Nephritic syndromes may present with more rapidly progressive renal failure when compared with nephrotic syndrome.1 The urinalysis is significant for hematuria with RBC casts, and variable levels of proteinuria (typically, less than 3.5 g/day is seen in nephritic syndrome).1
Review of the Data
Nephrotic Syndromes
Minimal change disease. MCD is more common in children than adults, and only accounts for 10% to 15% of glomerular disease cases in adults.3 It is associated with Hodgkin’s lymphoma, NSAID use, and allergic conditions. There usually is an absence of hypertension (HTN). There are no glomerular basement membrane abnormalities seen on light microscopy. Electron microscopy shows effacement of podocytes. On urinalysis, oval fat bodies are seen, which are characteristic of heavy proteinuria. Complement levels are normal. Steroids are first-line treatment, but in adults with relapses or steroid resistance, immunosuppressive agents have also been used.2
Focal segment glomerlosclerosis.
FSGS is the most common primary glomerular disorder in the United States and is the most common cause of nephrotic syndrome among blacks.4,5 It is associated with HIV (collapsing variant), parvovirus B19, heroin use, sickle-cell disease, obesity, chronic vesicoureteral reflux, and HTN.4,6 Sclerosis of segmental glomeruli is seen on light microscopy.