A48-year-old female was admitted to the hospital with atrial fibrillation. Her medical history was significant for systemic lupus erythematosus (SLE) diagnosed 28 years ago, with low dose prednisone treatment for the past 15 years.
In the past year, the patient had experienced progressive difficulty swallowing and often choked on her food, resulting in a 35-pound weight loss. She also mentioned that she had been bruising easily.
On examination, the patient had bruising around her eyes and a thick, enlarged tongue with clear marks of her teeth visible on the surface.
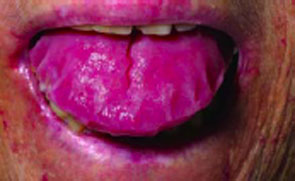
The 48-year-old patient’s enlarged tongue.
Which test will most likely lead to a diagnosis?
- Magnetic resonance imaging (MRI) of pituitary gland;
- Abdominal fat pad biopsy;
- Thyroid stimulating hormone (TSH) level;
- Serum Angiotensin converting enzyme (ACE) levels
- Tongue biopsy
Discussion
The answer is B: abdominal fat pad biopsy. Macroglossia, especially in adults, is a rare clinical finding and is most often associated with amyloidosis.1 It has also been rarely described in association with acromegaly, hypothyroidism, and sarcoidosis. In children, macroglossia may be seen in a variety of conditions including cretinism, Down syndrome, Beckwith-Wiedemann syndrome, and various storage diseases.2 Lymphangioma of the tongue may also present with macroglossia.3
Another finding classically seen in amyloidosis is palpebral purpura.4 The occurrence of these two physical findings together in this patient strongly suggests the diagnosis of amyloidosis. The amyloidoses are now considered a group of diseases characterized by extracellular deposition of insoluble fibrillar proteins in various organs secondary to misfolding of proteins.
They include not only primary and secondary amyloidosis but also Alzheimer’s, prion diseases, many other neurodegenerative disorders, and some types of cystic fibroses.5
Traditionally, amyloidosis was described as one of three types: primary, secondary, and heritable. Primary amyloidosis is the most common type and results from deposition of fragments of light chain immunoglobulin deposits. It is most frequently associated with plasma cell dyscrasias. Secondary, or AA, amyloidosis occurs in association with inflammatory conditions and results from deposition of fragments of the acute phase reactant serum amyloid A. Familial amyloidoses or the ATTR amyloidoses are a fairly heterogeneous group with different proteins associated with different disorders.5
The typical findings seen in this case are associated with primary amyloidosis.4 The presence of primary amyloidosis was confirmed in this case by a bone marrow biopsy. This patient had SLE, but SLE is usually not complicated by the development of amyloidosis, although rare cases in literature have been described.6 Also, as mentioned above, secondary amyloidosis is usually characterized by AA rather than AL amyloidosis.
The diagnosis of amyloidosis is made pathologically when an involved organ is biopsied. When clinical suspicion is high and no organ has been biopsied, the simplest procedure is to obtain an abdominal fat pad biopsy and stain it with Congo red. This test is 85% sensitive in patients with primary amyloidosis.4 TH
References
- Xavier SD, Bussoloti IF, Muller H. Macroglossia secondary to systemic amyloidosis: case report and literature review. Ear Nose Throat J. 2005 Jun;84(6):358-361.
- Wolford LM, Cottrell DA. Diagnosis of macroglossia and indications for reduction glossectomy. Am J Orthod Dentofacial Orthop. 1996 Aug;110:170-177.
- Gulemann M, Katz J. Macroglossia combined with lymphangioma: a case report. J Clin Pediatr Dent. 2003 Winter;27(2):167-169.
- Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997; 337: 898-909.
- Merlini G, Bellotti V. Mechanisms of disease: molecular mechanisms of amyloidosis. N Engl J Med. 2003; 349:583-596.
- Al-Hoqail I, Naddaf H, Al-Rikabi A, et al. Systemic lupus erythematosus and amyloidosis. Clin Rheumatol. 1997 Jun;16(4):422-424.on.