Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
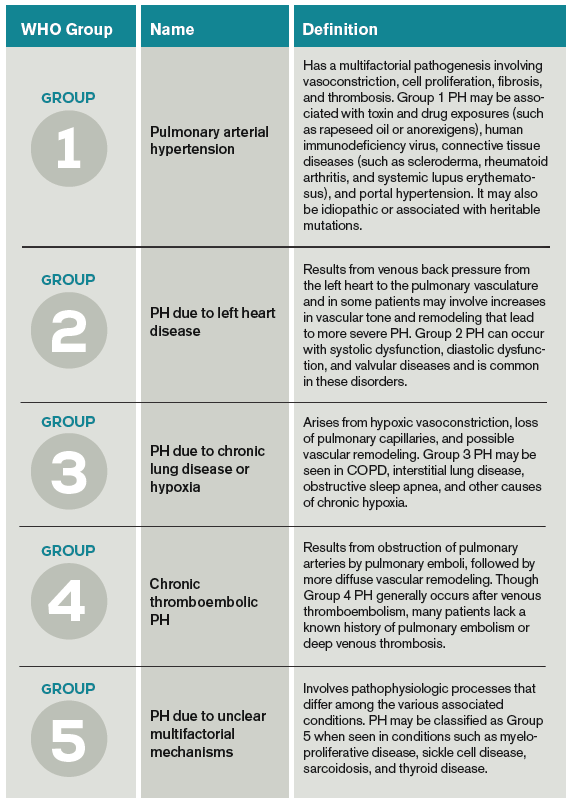
(Click for larger image.)Table 1. Basic pathophysiology of the five pulmonary hypertension (PH) WHO Groups and associated conditions.3,4,17
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).